Apalutamide
Chemical compound
From Wikipedia, the free encyclopedia
Apalutamide, sold under the brand name Erleada among others, is a nonsteroidal antiandrogen (NSAA) medication used for the treatment of prostate cancer.[2][8][9] It is an androgen receptor inhibitor.[2] It is taken by mouth.[2][10]

| |
 | |
| Clinical data | |
|---|---|
| Trade names | Erleada, others |
| Other names | ARN-509; JNJ-56021927; JNJ-927; A52 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a618018 |
| License data | |
| Pregnancy category |
|
| Routes of administration | By mouth[2] |
| Drug class | Nonsteroidal antiandrogen |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100%[2] |
| Protein binding | Apalutamide: 96%[2] NDMA: 95%[2] |
| Metabolism | Liver (CYP2C8, CYP3A4)[2] |
| Metabolites | • NDMA[2] |
| Elimination half-life | Apalutamide: 3–4 days (at steady-state)[7][2] |
| Excretion | Urine: 65%[2] Feces: 24%[2] |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.235.115 |
| Chemical and physical data | |
| Formula | C21H15F4N5O2S |
| Molar mass | 477.44 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
Side effects of apalutamide when added to castration include fatigue, nausea, abdominal pain, diarrhea, high blood pressure, rash, falls, bone fractures, and an underactive thyroid.[2][11][12][10][13] Rarely, it can cause seizures.[2][10] The medication has a high potential for drug interactions.[2][10] Apalutamide is an antiandrogen, and acts as an antagonist of the androgen receptor, the biological target of androgens like testosterone and dihydrotestosterone.[2][10][14] In doing so, it prevents the effects of these hormones in the prostate gland and elsewhere in the body.[2][10][14]
Apalutamide was first described in 2007, and was approved for the treatment of prostate cancer in February 2018.[8][9][10][15] It is the first medication to be approved specifically for the treatment of non-metastatic castration-resistant prostate cancer.[2][10][9]
Medical uses
Apalutamide is indicated for the treatment of people with metastatic castration-sensitive prostate cancer and the treatment of people with non-metastatic castration-resistant prostate cancer.[2][6]
Apalutamide[16] is used in conjunction with castration, either via bilateral orchiectomy or gonadotropin-releasing hormone analogue (GnRH analogue) therapy, as a method of androgen deprivation therapy in the treatment of non-metastatic castration-resistant prostate cancer.[2][17][18][19] It is also a promising potential treatment for metastatic castration-resistant prostate cancer (mCRPC), which the NSAA enzalutamide and the androgen synthesis inhibitor abiraterone acetate are used to treat.[13]
Contraindications
Contraindications of apalutamide include pregnancy and a history of or susceptibility to seizures.[2]
Side effects
Apalutamide has been found to be well tolerated in clinical trials,[20][17] with the most common side effects reported when added to surgical or medical castration including fatigue, nausea, abdominal pain, and diarrhea.[11][12][21] Other side effects have included rash, falls and bone fractures, and hypothyroidism, as well as seizures (in 0.2%), among others.[2][10][9] Apalutamide is an expected teratogen and has a theoretical risk of birth defects in male infants if taken by women during pregnancy.[2] It may impair male fertility.[2] When used as a monotherapy (i.e., without surgical or medical castration) in men, NSAAs are known to produce additional, estrogenic side effects like breast tenderness, gynecomastia, and feminization in general by increasing estradiol levels.[22] Similarly to the related second-generation NSAA enzalutamide but unlike first-generation NSAAs like flutamide and bicalutamide, elevated liver enzymes and hepatotoxicity have not been reported with apalutamide.[2] Case reports of rare interstitial lung disease with apalutamide exist similarly to with first-generation NSAAs however.[23][24][25]
Overdose
Interactions
Apalutamide has a high potential for drug interactions.[2] In terms of effects of apalutamide on other drugs, the exposure of substrates of CYP3A4, CYP2C19, CYP2C9, UDP-glucuronosyltransferase, P-glycoprotein, ABCG2, or OATP1B1 may be reduced to varying extents.[2] In terms of effects of other drugs on apalutamide, strong CYP2C8 or CYP3A4 inhibitors may increase levels of apalutamide or its major active metabolite N-desmethylapalutamide, while mild to moderate CYP2C8 or CYP3A4 inhibitors are not expected to affect their exposure.[2]
Pharmacology
Pharmacodynamics
Antiandrogenic activity
Apalutamide acts as a selective competitive silent antagonist of the androgen receptor (AR), via the ligand-binding domain, and hence is an antiandrogen.[10][14][11][17] It is similar both structurally and pharmacologically to the second-generation NSAA enzalutamide,[20][26] but shows some advantages, including higher antiandrogenic activity as well as several-fold reduced central nervous system distribution.[14][11][17] The latter difference may reduce its comparative risk of seizures and other central side effects.[14][11][17] Apalutamide has 5- to 10-fold greater affinity for the AR than bicalutamide, a first-generation NSAA.[19][18]
The acquired F876L mutation of the AR identified in advanced prostate cancer cells has been found to confer resistance to both enzalutamide and apalutamide.[27][28] A newer NSAA, darolutamide, is not affected by this mutation, nor has it been found to be affected by any other tested/well-known AR mutations.[29] Apalutamide may be effective in a subset of prostate cancer patients with acquired resistance to abiraterone acetate.[20]
Other activities
Apalutamide shows potent induction potential of cytochrome P450 enzymes similarly to enzalutamide.[2][30][31] It is a strong inducer of CYP3A4 and CYP2C19 and a weak inducer of CYP2C9, as well as an inducer of UDP-glucuronosyltransferase.[2] In addition, apalutamide is an inducer of P-glycoprotein, ABCG2, and OATP1B1.[2]
Apalutamide binds weakly to and inhibits the GABAA receptor in vitro similarly to enzalutamide (IC50 = 3.0 and 2.7 μM, respectively),[14] but due to its relatively lower central concentrations, may have a lower risk of seizures in comparison.[14][11][21]
Apalutamide has been found to significantly and concentration-dependently increase QT interval.[2]
Pharmacokinetics
The mean absolute oral bioavailability of apalutamide is 100%.[2] Mean peak levels of apalutamide occur 2 hours following administration, with a range of 1 to 5 hours.[2] Food delays the median time to peak levels of apalutamide by approximately 2 hours, with no significant changes in the peak levels themselves or in area-under-curve levels.[2] Steady-state levels of apalutamide are achieved following 4 weeks of administration, with an approximate 5-fold accumulation.[2] Peak concentrations for 160 mg/day apalutamide at steady-state are 6.0 μg/mL (12.5 μmol/L),[2] relative to peak levels of 16.6 μg/mL (35.7 μmol/L) for 160 mg/day enzalutamide and mean (R)-bicalutamide levels of 21.6 μg/mL (50.2 μmol/L) for 150 mg/day bicalutamide.[32][33] The mean volume of distribution of apalutamide at steady-state is approximately 276 L.[2] The plasma protein binding of apalutamide is 96%, while that of its major metabolite N-desmethylapalutamide is 95%, both irrespective of concentration.[2]
Apalutamide is metabolized in the liver by CYP2C8 and CYP3A4.[2] A major active metabolite, N-desmethylapalutamide, is formed by these enzymes, with similar contribution of each of these enzymes to its formation at steady-state.[2] Following a single oral dose of 200 mg apalutamide, apalutamide represented 45% and N-desmethylapalutamide 44% of total area-under-curve levels.[2] The mean elimination half-life of apalutamide at steady-state is 3 to 4 days.[2][7] Fluctuations in apalutamide exposure are low and levels are stable throughout the day, with mean peak-to-trough ratios of 1.63 for apalutamide and 1.27–1.3 for N-desmethylapalutamide.[2] After a single dose of apalutamide, its clearance rate (CL/F) was 1.3 L/h, while its clearance rate increased to 2.0 L/h at steady-state.[10] This change is considered to be likely due to CYP3A4 auto-induction.[10] Approximately 65% of apalutamide is excreted in urine (1.2% as unchanged apalutamide and 2.7% as N-desmethylapalutamide) while 24% is excreted in feces (1.5% as unchanged apalutamide and 2% as N-desmethylapalutamide).[2]
Chemistry
Apalutamide is a structural analogue of enzalutamide and RD-162.[19][34] It is a pyridyl variant of RD-162. Enzalutamide and RD-162 were derived from the nonsteroidal androgen RU-59063, which itself was derived from the first-generation NSAA nilutamide and by extension from flutamide.[35]

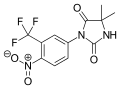



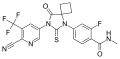 Apalutamide
Apalutamide





Synthesis
The chemical synthesis of apalutamide has been described.[36][37] Also see:[38]

The bromination of 2-hydroxy-5-nitro-3-(trifluoromethyl)pyridine [99368-66-8] (1) with POBr3 gave 2-bromo-5-nitro-3-(trifluoromethyl)pyridine [956104-42-0] (2). A cyanation using sodium cyanide and copper(I)iodide in butyronitrile provided 5-nitro-3-trifluoromethylpyridine-2-carbonitrile [573762-57-9] (3). Chemoselective reduction of the nitro group involved a catalyst slurry (H3PO2, Pt/C, NH4VO3) in xylenes and butyronitrile under a hydrogen atmosphere to give 5-amino-3-(trifluoromethyl)pyridine-2-carbonitrile [573762-62-6] (4). A telescoped CDI mediated coupling reaction with Boc-cyclovaline [120728-10-1] (5) yielded amide, PC121422675 (6). Boc cleavage using HCl in IPA gave 1-amino-N-[6-cyano-5-(trifluoromethyl)-3-pyridyl]cyclobutanecarboxamide [1950587-17-3] (7). An Ullman-type coupling with 4-bromo-2-fluoro-N-methylbenzamide [749927-69-3] (8) gave [1950587-20-8] (9). Lastly, exposure to 1,1'-thiocarbonyldi-2(1H)-pyridone [102368-13-8] (10) and DMAP in warm DMA provided apalutamide (11).
History
Apalutamide was originated by the University of California system and was developed primarily by Janssen Research & Development, a division of Johnson & Johnson.[39] It was first described in the literature in a United States patent application that was published in November 2007 and in another that was submitted in July 2010.[15][40] A March 2012 publication described the discovery and development of apalutamide.[14] A phase I clinical trial of apalutamide was completed by March 2012, and the results of this study were published in 2013.[14][41] Information on phase III clinical studies, including ATLAS, SPARTAN, and TITAN, was published between 2014 and 2016.[42][43][44] Positive results for phase III trials were first described in 2017, and Janssen submitted a New Drug Application for apalutamide to the United States Food and Drug Administration on 11 October 2017.[45] Apalutamide was approved by the Food and Drug Administration in the United States, under the brand name Erleada, for the treatment of non-metastatic castration-resistant prostate cancer in February 2018.[8][9] It was subsequently approved in Canada, the European Union, and Australia.[46][6]
Society and culture
Generic names
Apalutamide is the generic name of the medication and is its international nonproprietary name.[47][46] It is also known by its developmental code names ARN-509 and JNJ-56021927.[39][10]
Brand names
Apalutamide is marketed under the brand names Erleada and Erlyand.[2][8][9][46]
Availability
Apalutamide is available in the United States, Canada, the European Union, and Australia.[2][8][9][46][6]